진단명보다 중요한 것 – 기전(mechanism)으로 이해하는 CMT
📌 Series Introduction
이 시리즈는 AI 기반 유전변이 판독 도구가 보편화된 시대에, 자동화된 우선순위화 이후 판독자가 실제로 무엇을 이해하고 어떻게 판단해야 하는지에 초점을 맞춥니다.
각 편에서는 하나의 질환군을 중심으로, 유전적 기전과 임상 스펙트럼을 함께 살펴보며 변이를 “찾는 것”을 넘어 “설명하고 해석하는” 데 필요한 관점을 공유하고자 합니다.
Episode 1 — Charcot–Marie–Tooth Disease (CMT)
왜 첫 번째 질환으로 CMT인가
Charcot–Marie–Tooth disease (CMT)는 가장 흔한 유전성 말초신경병증 중 하나입니다.
하지만 “흔하다”는 인상과 달리, 실제 유전변이 판독 과정에서는 가장 많은 오해와 혼란이 발생하는 질환군이기도 합니다.
CMT는 하나의 질환명이지만, 그 안에는 서로 다른 유전자, 서로 다른 표현형, 서로 다른 임상 경과가 공존하기도 합니다.
따라서 “CMT 관련 유전자에 변이가 있다”는 사실만으로는 환자의 증상을 충분히 설명하기 어려운 경우가 있습니다.
CMT는 단순히 ‘이름’으로 이해하기보다는, 각 유전자가 만들어내는 분자적 기전과 표현형을 함께 고려하며 폭넓고 깊게 접근해야 하는 대표적인 질환군입니다.”
이러한 이유로 CMT는, AI 기반 우선순위화 이후 판독자가 반드시 ‘기전 중심 사고’로 돌아와야 하는 대표적인 질환군이라 할 수 있습니다.
Charcot–Marie–Tooth Disease란 무엇인가
Charcot–Marie–Tooth disease(CMT)는 가장 흔한 유전성 말초신경병증이지만, 임상과 유전 판독 모두에서 가장 복잡한 질환군 중 하나이기도 합니다.
임상적으로는 팔과 다리의 원위부(distal)에서 증상이 시작되는 것이 특징이며, 질환이 진행됨에 따라 근육 위축, 보행 이상, 감각 저하가 점차 뚜렷해지며 아래와 같은 임상 특징을 보입니다.
CMT의 핵심 임상 특징
- Slowly progressive distal motor neuropathy
- Distal muscle weakness and atrophy
- Pes cavus(요족) 변형
- Tendon reflex 감소 또는 소실
- 발병 시기: 주로 10–30세 (영아기~고령 발병 가능)
- 유병률: 약 1/2,500
- 주요 병변: Peripheral Nervous System (PNS)
CMT를 이해하는 출발점: 말초신경의 구조
말초 신경은 크게 두 가지 요소로 구성됩니다. 축삭(axon)은 신경 신호를 전달하는 중심구조이고,
이를 둘러싼 수초(myelin)는 신호 전달 속도를 높이기 위해 도약전도(saltatory conduction)를 가능하게 합니다.
CMT는 이 구조 중
- 수초에 문제가 생기는 경우와
- 축삭 자체에 문제가 생기는 경우로 나뉘며,
이 구조적 차이는 단순한 분류 문제가 아니라, 이후 유전자 선택과 판독 전략 전체를 좌우합니다.
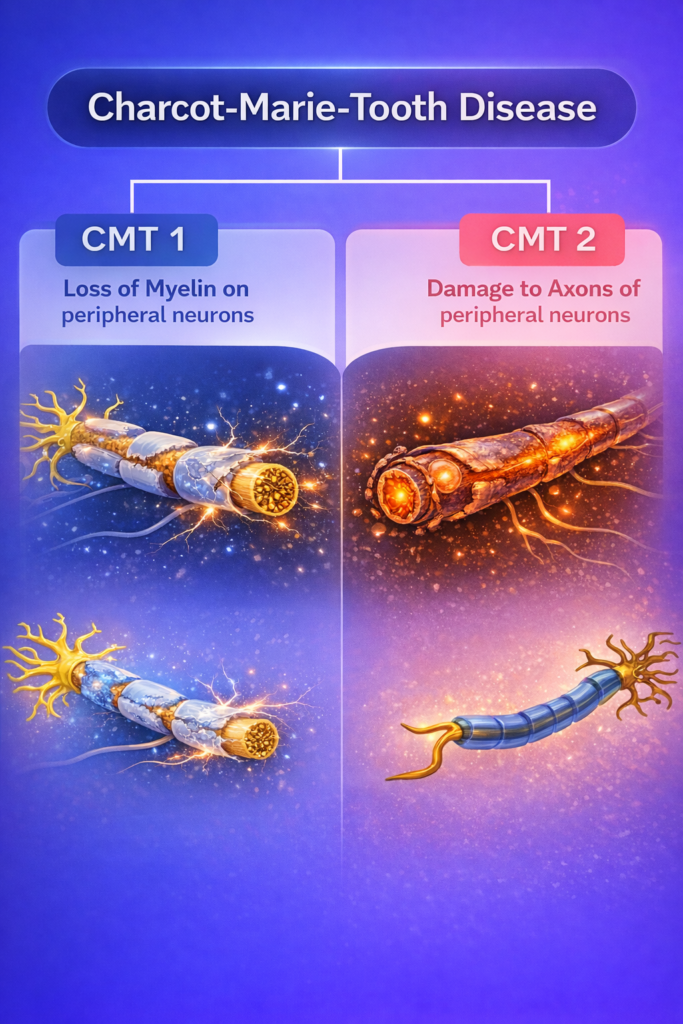
Demyelinating CMT vs Axonal CMT
Demyelinating CMT
- 수초의 구조 또는 유지 실패
- Nerve Conduction Velocity (NCV) 현저히 감소
- 경우에 따라 신경 비대(nerve enlargement) 동반
Axonal CMT
- 축삭의 구조적·기능적 손상
- NCV는 비교적 유지되나 amplitude 감소
- 길이에 비례한(length-dependent) 신경 손상
임상 증상만으로는 두 타입을 명확히 구분하기 어렵기 때문에, 전기생리 검사(NCV, amplitude) 정보는 CMT 판독에서 매우 중요한 분기점이 됩니다.
CMT 판독의 핵심: 대표 유전자와 기전
CMT에는 수십 개 이상의 유전자가 관여하지만, 판독 관점에서는 질환의 병태생리를 대표적으로 보여주는 유전자들을 기전 중심으로 이해하는 접근이 중요합니다.
아래에서는 CMT 판독에서 기전 이해의 기준점이 되는 대표 유전자들을, 각각의 병적 메커니즘 중심으로 살펴봅니다.
1) PMP22 — demyelinating CMT의 대표적인 예시
PMP22는 demyelinating CMT를 대표하는 핵심 유전자입니다.
Schwann cell에서 발현되는 myelin 단백질로, 수초의 형성과 안정성 유지에 중요한 역할을 합니다.
PMP22의 특징은 변이의 종류에 따라 병적 기전이 완전히 달라진다는 점입니다.
- Duplication
→ PMP22 과발현
→ proteasome overload
→ intracellular aggregates
→ Schwann cell 성숙 장애
→ 전형적인 CMT1A
- Point mutation
→ 단백질의 세포막 이동 실패
→ ER stress 증가
→ demyelination
- Deletion
→ 수초 불안정성 증가
→ 압박에 취약한 신경
→ HNPP (Hereditary Neuropathy with liability to Pressure Palsies)
PMP22는
“변이가 있느냐”보다 변이의 유형(예: dosage 변화 또는 point mutation)과 그에 따른 분자적 발병 기전을 함께 고려해야 합니다.
2) MPZ & SH3TC2 — 수초 구조와 유지 실패
Demyelinating CMT에서 PMP22 다음으로 반드시 함께 고려해야 할 유전자들이 MPZ와 SH3TC2입니다. 이들은 수초의 “양”이 아니라 구조적 완성도와 유지 능력에 관여합니다.
MPZ — 수초 구조를 지탱하는 핵심 단백질
MPZ(Myelin Protein Zero, P0)는 말초 수초 단백질의 약 50%를 차지하는 가장 중요한 구조 단백질이며 수초 층 사이의 adhesion을 유지하는 역할을 합니다.
- 병적 기전
→ myelin adhesion 실패
→ 수초 구조 붕괴
→ demyelination
따라서 MPZ 관련 CMT에서는 단순한 탈수초 여부보다, 수초 구조 붕괴라는 병적 기전을 중심으로 판독하는 접근이 필요합니다.
3)SH3TC2 — Node of Ranvier의 형성에 필수적인 단백질
SH3TC2는 endosomal recycling과 myelination 조절에 관여하며, NRG1/ErbB signaling과 밀접하게 연결되어 있습니다.
- 병적 기전
→ NRG1/ErbB signaling 장애
→ hypomyelination
→ 반복적 탈수초–재수초
→ onion bulb formation
이 두 유전자는 “demyelinating”이라는 분류 아래 묶이지만, 병태생리는 서로 다르다는 점이 중요합니다.
4) GJB1 — gap junction 장애와 CNS involvement
GJB1은 Connexin 32 단백질을 생성하며, gap junction을 형성하고 Schwann cell–axon 간 대사적 교환(metabolic coupling)을 유지합니다.
또한 oligodendrocyte에서도 발현되기 때문에, CMT 유전자 중 드물게 중추신경계 증상을 동반할 수 있습니다.
- Loss of function
→ 정상적인 channel 형성 불가
→ cell surface로 이동 불가
→ gap junction 기능 소실
→ myelin 손상
→ 이차적 axonal degeneration
- Gain of function
→ Hemichannel 결합 실패
→ 비정상적 ion leakage
→ Intracellular Ion homeostasis 파괴
→ 더 심한 신경 손상
→ CNS 증상 동반 가능
GJB1 변이가 확인된 경우에는 말초 신경 증상뿐 아니라 CNS involvement 여부도 함께 고려해야 합니다.
5) MFN2 and GDAP1 — 미토콘드리아 기능 이상과 축삭성 CMT
MFN2는 axonal CMT를 대표하는 유전자입니다. 미토콘드리아 fusion과 네트워크 유지에 관여하며, 긴 축삭을 가진 말초 신경에서 에너지 공급에 핵심적인 역할을 합니다.
- 병적 기전
→ 미토콘드리아 fusion 장애
→ ER–mitochondria 연결 이상
→ 축삭 말단 에너지 공급 실패
GDAP는 Ganglioside-induced differentiation-associated protein 1을 생성합니다.
- 병적 기전
→미토콘드리아 fusion/fission 균형 붕괴
→비정상적인 미토콘드리아 분포 및 기능 장애 유발 - 특징적인 임상 소견
→ GDAP1 관련 환자에서는
성대 마비(vocal cord paralysis) 쉰 목소리(hoarseness)와 같은 매우 독특한 증상이 동반될 수 있음
MFN2 관련 CMT는 NCV는 비교적 보존되지만 amplitude 감소가 두드러지는 경우가 많고, optic atrophy, hearing loss 등 확장된 임상 증상을 동반할 수 있습니다.
6) HINT1 — mRNA processing 장애에 의해 유발되는 대사성 축삭 신경병증
HINT1은 purine phosphoramidase 효소로 작용하며, homodimer 형태로 기능합니다.
단순한 대사 효소에 그치지 않고, 전사 인자 활성 조절과 mRNA processing pathway에도 관여합니다.
또한 μ-opioid receptor(MOR)와 상호작용하여 통증 및 감각 신호 조절에 기여합니다.
- 병적 기전
→ 효소 활성 부위(active site)의 결함
→ 단백질 불안정성 증가 및 nonsense-mediated decay (NMD) 발생
→ dimerization 장애
→ mRNA processing pathway 붕괴
→ 대사 불균형으로 인한 축삭 퇴행(axonal degeneration)
HINT1는 “유전자 기능 분류”만으로는 설명되지 않으며, 기전 중심 해석이 왜 필요한지를 잘 보여주는 대표적인 예입니다.
7) SORD — 대사성 축삭 신경병증과 NGS 해석의 함정
SORD는 polyol pathway의 핵심 효소로, glucose → sorbitol → fructose 대사 과정에 관여합니다.
- 병적 기전
→ sorbitol 축적
→ osmotic / oxidative stress
→ NADPH 고갈
→ 축삭 손상
SORD는 NGS 해석 시 기술적 주의점이 매우 중요한 유전자입니다.
- SORD2P pseudogene 존재 (서열 유사도 ~90%)
- 대표 변이 c.757delG는 pseudogene에서는 정상 서열
따라서 SORD 변이는 coverage와 read alignment를 반드시 확인해야 합니다.
CMT는 말초 신경에만 머무르지 않는다
CMT는 유전자에 따라 다양한 전신 증상을 동반할 수 있습니다.
- Hearing Loss (GJB1, PMP22, NEFL)
- Optic Atrophy (MFN2)
- Vocal Cord Paralysis (GDAP1, TRPV4)
- Cognitive Decline (MFN2, MCM3AP)
- Transient Encephalopathy (GJB1)
- Renal Failrue (INF2)
- Scoliosis (SH3TC2)
- Neuromyotonia (HINT1)
이러한 전신 증상은 종종 CMT와 직접 연결되지 않은 채 별개의 문제로 인식되기 쉽습니다.
그 결과, 표현형–유전자 연결이 끊기면서 실제 원인 변이가 판독 과정에서 배제되는 경우가 적지 않습니다.
CMT 판독에서 자주 생기는 오해 TOP 5
1️⃣ CMT는 하나의 질환이다
2️⃣ 임상 증상만으로 subtype을 구분할 수 있다
3️⃣ CMT는 말초 신경 증상만 나타난다
4️⃣ 병적 변이가 있으면 바로 진단이 가능하다
5️⃣ AI 우선순위가 높으면 원인변이일 가능성도 높다
→이 오해들은 대부분, ‘variant 결과만을 보고 해석을 시작할 때’ 발생합니다.
CMT 판독 시 추가로 유의할 점
1️⃣ CNV를 반드시 함께 고려해야 한다
CMT는 single nucleotide variant (SNV)과 더불어 copy number variant (CNV)가 원인인 경우가 매우 많은 질환군입니다.
- PMP22 duplication / deletion은 CMT 판독에서 가장 흔한 원인 중 하나
- 단순 SNV 중심 분석만으로는 대표적인 CMT1A / HNPP 케이스를 놓칠 수 있습니다
👉 WES/WGS 판독 시 CNV caller 결과를 반드시 함께 확인해야 합니다.
2️⃣ Inheritance mode를 먼저 고정하지 말 것
CMT는 AD / AR / X-linked 유전 양식이 모두 가능한 질환군입니다.
- 초기 임상 정보만 보고 AD CMT로 단정하는 경우가 많지만 실제로는 AR-CMT, X-linked CMT (GJB1)도 흔합니다.
👉 판독 초기 단계에서 유전 양식을 미리 제한하면 후보 변이를 스스로 좁혀버리는 오류가 발생할 수 있습니다.
3️⃣ Early-onset vs late-onset을 지나치게 단순화하지 말 것
전통적으로는
- early-onset → demyelinating
- late-onset → axonal
로 구분해왔지만,
실제 임상에서는
- MFN2 early-onset axonal CMT
- MPZ late-onset demyelinating CMT
처럼 예외가 매우 많습니다.
👉 발병 시기는 참고 정보이지, subtype 결정 기준은 아닙니다.
4️⃣ Electrophysiology 결과를 ‘정량적’으로 볼 것
NCV 감소 / amplitude 감소를 단순히 “감소했다”로 표현하면 해석이 흐려집니다.
- NCV < 38 m/s → demyelinating 가능성 높음
- amplitude 감소 + NCV 보존 → axonal 가능성
👉 수치 기반으로 해석해야 유전자–기전 연결이 명확해집니다.
5️⃣ Phenotype 확장을 의도적으로 열어둘 것
CMT는 단순 말초신경병증으로 시작하지만, 판독 과정에서는 표현형 확장을 일부러 열어두는 태도가 필요합니다.
예:
- optic atrophy → MFN2
- vocal cord paralysis → GDAP1
- transient CNS symptoms → GJB1
- neuromyotonia → HINT1
👉 초기 HPO 세트가 너무 좁으면 진짜 원인 유전자를 배제해버리는 경우가 발생합니다.
6️⃣ “CMT-like phenotype”도 염두에 둘 것
모든 말초신경병증이 CMT는 아닙니다.
- Hereditary sensory neuropathy
- Distal hereditary motor neuropathy (dHMN)
- Mitochondrial disease
- Metabolic neuropathy
👉 CMT panel 음성일 때, “CMT가 아닌 다른 기전의 말초신경병증” 가능성도 함께 고려해야 합니다.
7️⃣ Negative result도 의미가 있다
CMT는 아직도
- 원인 유전자가 밝혀지지 않은 케이스
- 구조 변이/비정형 변이가 원인인 케이스
가 적지 않습니다.
👉 “negative”는 실패가 아니라, 다음 접근(WGS, reanalysis, long-read 등)을 결정하는 정보입니다.
정리하며: CMT 판독이 주는 메시지
CMT는 유전변이 판독에서 gene ≠ diagnosis, variant ≠ causative라는 원칙을 가장 분명하게 보여주는 질환군입니다.
정확한 판독을 위해서는 유전자 이름을 나열하는 수준을 넘어, 신경 생물학적 경로 중 어느 단계에서 문제가 발생했는지 파악해야 합니다.
그리고 이것이 바로, GEBRA와 같은 AI 기반 도구가 아무리 발전하더라도 판독자의 역할이 여전히 중요한 이유입니다.
답글 남기기
댓글을 달기 위해서는 로그인해야합니다.