📌 Series Introduction
In an era where AI-based variant interpretation tools have become widely adopted, this series focuses on what human interpreters must still understand and decide after automated prioritization.
Each episode centers on a specific disease group, examining genetic mechanisms and clinical spectra together, with the goal of moving beyond simply “finding variants” toward explaining and interpreting them in a clinically meaningful way.
Key Takeaway
ALS is not simply a motor neuron disease.
👉 It is a disease where genes + cellular homeostasis + neural networks collapse simultaneously
AI can now effectively prioritize variants.
However, in diseases like ALS, what comes after that is far more important.
- Which biological pathway is this gene involved in?
- How does this variant actually connect to the patient’s phenotype?
- Where does this patient lie on the ALS–FTD spectrum?
And the one who answers these questions is not AI, but the interpreter.
How should we understand ALS?
Amyotrophic Lateral Sclerosis (ALS) is often described as:
“a disease where muscles gradually weaken.”
But in reality, ALS is far more complex.
👉 ALS is a disease where the entire system controlling movement collapses.
1. ALS is not a “simple progressive disease”
ALS is a progressive disease where muscle function declines over time.
Diagnosis can take months to years, and there is currently no curative treatment.
There is also a common misconception:
No family history means it is not genetic? → False
ALS:
- Often appears sporadic
- But frequently has underlying genetic contributions
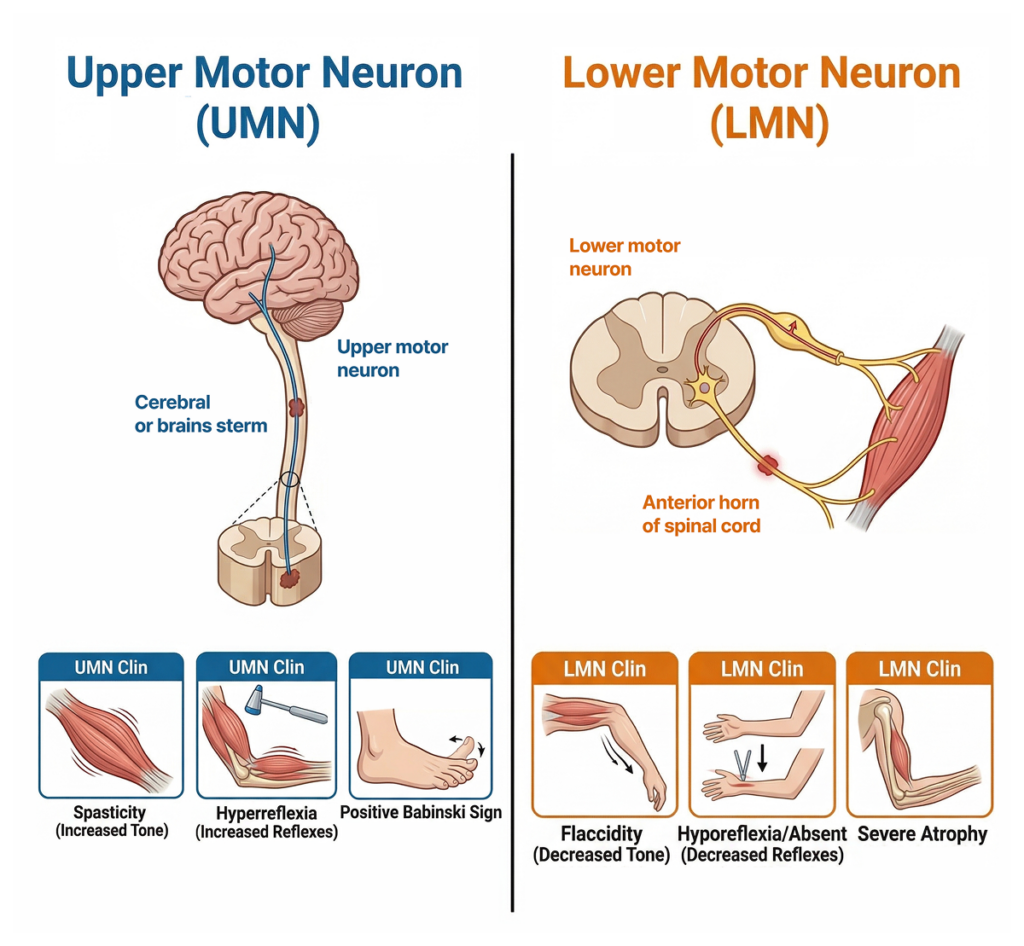
2. Core of ALS: UMN + LMN collapse together
The most important point in understanding ALS is this:
👉 Upper Motor Neuron (UMN) + Lower Motor Neuron (LMN) are both affected simultaneously
Why does this matter?
UMN damage
- Spasticity
- Hyperreflexia
- Positive Babinski sign
LMN damage
- Muscle atrophy
- Fasciculations
- Decreased muscle tone
Most diseases affect only one axis.
👉 ALS affects both axes simultaneously
Additionally, in some patients:
👉 Frontal-temporal dysfunction (cognitive/behavioral changes) may occur
(Up to 45% of ALS patients show cognitive impairment)
3. ALS is not diagnosed with a single test
ALS diagnosis is not based on a single test.
👉 Clinical findings + EMG + pattern of involvement + progression
It is defined stepwise:
- suspected → possible → probable → definite ALS
Key point:
👉 “Are UMN + LMN signs consistently observed across multiple anatomical regions?”
4. ALS-like but not ALS diseases
Some diseases resemble ALS but are different.
Examples:
- SBMA (AR)
- SMA type IV
- APBD (GBE1)
- HEXA-related disorders
Common feature:
👉 They are biased toward a single axis
Whereas:
👉 ALS = UMN + LMN together
👉 Others = biased toward UMN, LMN, or non-motor features
5. Sporadic vs Familial ALS
ALS is usually classified as:
Familial ALS (fALS)
- Strong single-gene influence
Sporadic ALS (sALS)
- Environment + aging + polygenic factors
However, clinically:
👉 Pathogenic variants can be found even in seemingly sporadic cases
👉 Negative family history ≠ absence of genetic cause
6. Key ALS-related genes
Major genes to consider:
- C9orf72
→ Most common genetic cause, strongly linked to FTD
→ Autosomal dominant
→ 39–45% of familial ALS - SOD1
→ One of the earliest known ALS genes
→ AD (some AR)
→ 15–20% - FUS
→ Early onset (<51 years)
→ AD
→ 4–8% - TARDBP (TDP-43)
→ Average onset ~53.5 years
→ AD
→ 1–4%
Other genes:
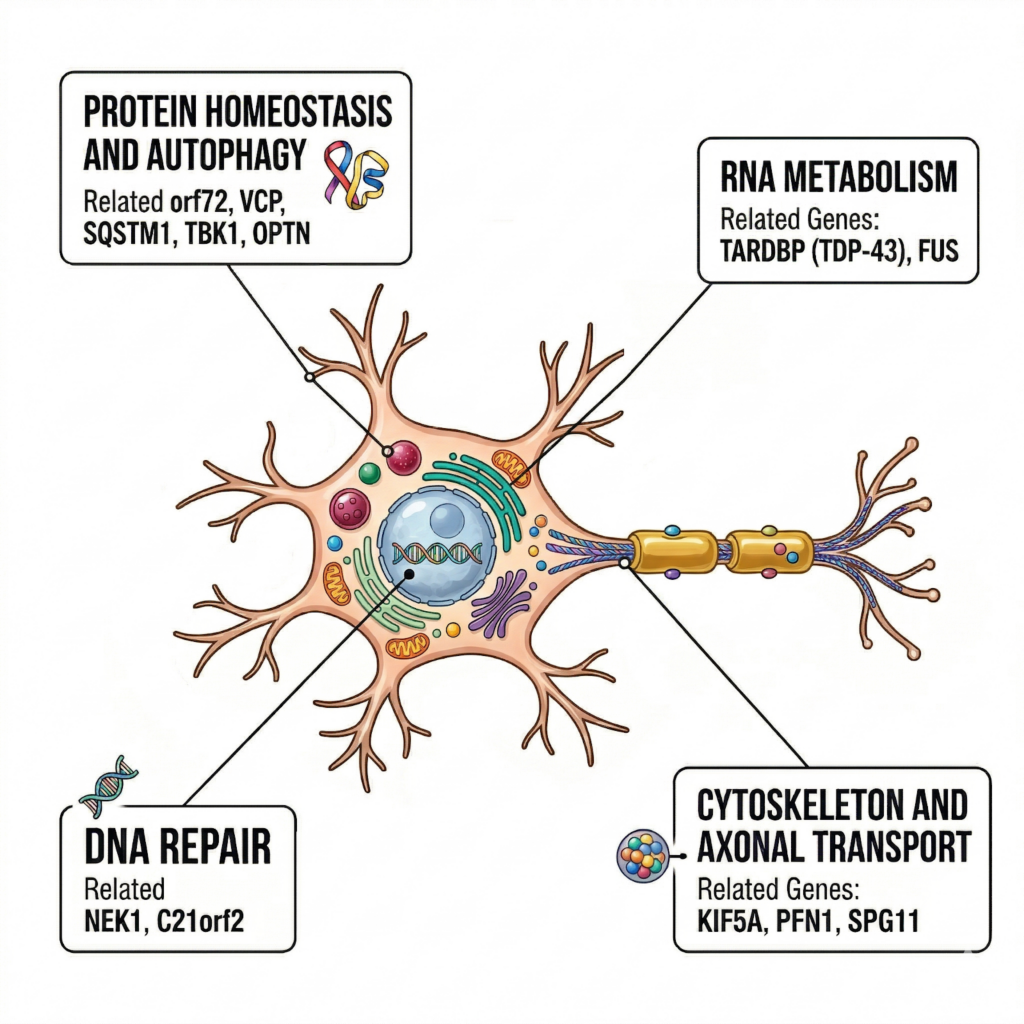
VCP, TBK1, OPTN, SQSTM1, CHCHD10, KIF5A, NEK1
Functions include:
- Proteostasis / autophagy
- RNA metabolism
- DNA repair
- Cytoskeleton & axonal transport
7. ALS is connected to FTD
This is often overlooked in interpretation.
ALS and FTD are not separate diseases.
👉 They are part of a spectrum
- Pure ALS (motor)
- FTD (cognitive/behavioral)
- ALS–FTD (both)
👉 Cognitive changes are not exceptions—they are part of the disease spectrum
8. Genes reveal the nature of ALS
ALS genes do not share a single common pathway.
Because ALS is not a single-pathway disease.
Key axes:
- Proteostasis / autophagy
- RNA metabolism
- DNA repair
- Cytoskeleton & transport
👉 ALS = collapse of the entire neuronal maintenance system
9. What ultimately fails?
All pathways converge to:
Mitochondrial dysfunction
ER stress & UPR activation
Ca²⁺ signaling disruption
Result:
👉 Energy failure
👉 Protein handling defects
👉 Increased cellular stress
→ leading to motor neuron loss
Why this matters for interpretation
AI can now find variants effectively.
The real problem comes next.
In ALS-like cases, interpretation must be structured across three layers:
1. Gene-level context
→ Which biological pathway does this gene belong to?
→ Does it match known ALS mechanisms?
2. Phenotype matching
→ Are both UMN + LMN involved?
→ Any cognitive/behavioral features suggesting ALS–FTD?
3. Spectrum positioning
→ Where does this case lie on the ALS–FTD continuum?
→ Can the variant explain both motor and non-motor features?
This process goes beyond classification.
It connects layers of information to explain the patient.
In real workflows, reasoning must flow naturally across:
- gene function
- phenotype
- prior interpretations
👉 AI performs prioritization
👉 The interpreter builds reasoning
This series exists for that purpose:
Not how to better “find” variants,
but how to go beyond them —
and clearly structure how we explain the patient.
👉 See how this reasoning applies in real cases
→ GEBRA page
References
Nijs, M. & Van Damme, P. The genetics of amyotrophic lateral sclerosis. Curr. Opin. Neurol. 37, 560–569 (2024).
Siddique, N. & Siddique, T. Amyotrophic Lateral Sclerosis Overview. GeneReviews® (2001; updated 2023).
답글 남기기
댓글을 달기 위해서는 로그인해야합니다.