
📍Key Takeaways
- EDS is more common than it appears — with an estimated prevalence of at least 1 in 5,000, there may already be undiagnosed patients in your clinic.
- Patients with EDS wait an average of over 10 years for a confirmed diagnosis, often after being told repeatedly that “nothing is wrong.”
- When joint hypermobility, skin abnormalities, and unexplained chronic pain occur together, EDS belongs on your differential — and genetic testing can confirm the subtype.

What Is EDS?
Ehlers-Danlos syndrome (EDS) is a group of heritable connective tissue disorders caused by defects in collagen synthesis or structure. Because connective tissue is distributed throughout the body — in joints, skin, blood vessels, and internal organs — symptoms can manifest across multiple organ systems simultaneously.
The 2017 International EDS Classification (Malfait et al.) recognizes 13 subtypes involving more than 19 genes. Subtype identification is the essential first step in guiding clinical management, as presentation and prognosis differ substantially. The three subtypes most likely to be encountered in clinical practice are:
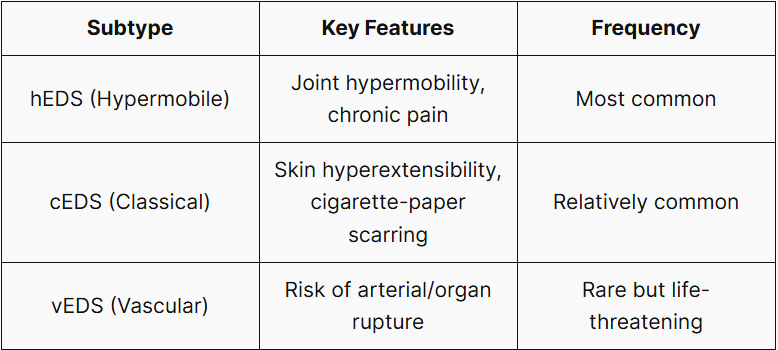
More Common Than You Think
The overall prevalence of EDS is estimated at a minimum of 1 in 5,000 (conservative estimate). The most common subtype, hEDS, was reported at a diagnosed prevalence of approximately 1 in 515 in a 2019 nationwide electronic cohort study in Wales, UK (Demmler et al., BMJ Open) — substantially higher than prior estimates, underscoring an urgent need for greater clinical awareness.
Given the well-documented delays in diagnosis, the true prevalence is likely higher still. EDS may already be in your clinic.
Clinical Features
EDS symptoms fall into three main categories:
① Joints
- Hypermobility beyond the normal range of motion
- Recurrent dislocations or subluxations
- Unexplained chronic joint pain
② Skin
- Abnormally hyperextensible or velvety-soft skin
- Wide, atrophic “cigarette-paper” scarring following minor trauma
- Easy bruising
③ Systemic / Other
- Unexplained chronic pain and fatigue
- Autonomic dysfunction: postural orthostatic tachycardia syndrome (POTS), syncope
- Gastrointestinal involvement (gastroparesis, IBS-like symptoms)
⚠️ vEDS is different. Vascular EDS carries a risk of spontaneous arterial or visceral rupture, with a reported median survival age of approximately 51 years. Because musculoskeletal and skin findings may be subtle or absent, vEDS is easily confused with other subtypes. Early identification through family history evaluation and genetic testing is critical.
Why Is It So Often Missed?
Patients with EDS wait an average of more than 10 years for a confirmed diagnosis, often moving between multiple specialties in the interim.
The delay is largely structural:
- Symptoms span multiple specialties → each clinician independently finds “nothing wrong”
- Fatigue and pain are frequently attributed to psychological causes
- Common misdiagnoses: fibromyalgia, chronic fatigue syndrome, irritable bowel syndrome
When a patient presents with multiple unexplained complaints and a history of repeated specialist visits, EDS deserves a place in the differential diagnosis.
When to Suspect EDS
EDS may be worth considering when the following features occur in combination.
- Recurrent joint dislocations or hypermobility
- Abnormally stretchy or velvety skin
- Wide, atrophic scarring after minor trauma
- Unexplained chronic musculoskeletal pain
- Tachycardia on standing, syncope
- Family history of similar symptoms
Formal diagnosis follows the 2017 International EDS Classification Criteria, including Beighton Score assessment.
Confirming the Diagnosis

hEDS can be diagnosed clinically using the 2017 diagnostic criteria, which incorporate the Beighton Score for joint hypermobility assessment. For all other subtypes — and critically for vEDS — genetic testing is required to confirm the subtype and exclude life-threatening variants.
Genetic testing is particularly recommended when:
- Family history of vascular complications — vEDS follows an autosomal dominant inheritance pattern, meaning genetic testing can identify at-risk family members before a vascular event occurs
- Subtype clarification needed to guide management — COL3A1 variant type predicts the pattern of arterial involvement and informs surgical planning; confirmed diagnosis is associated with significantly better operative outcomes
Early identification enables targeted management, complication prevention, and improved quality of life. If EDS is on your differential, genetic testing can confirm the diagnosis. Find out about pricing, process, and more via the button below.
Want to learn more about EDS?
*This post was produced in recognition of May as EDS Awareness Month, with the goal of spreading awareness about EDS and shortening the long diagnostic odyssey that patients face.
References
- Malfait F, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017. https://doi.org/10.1002/ajmg.c.31552
- Demmler JC, et al. Diagnosed prevalence of Ehlers-Danlos syndrome and hypermobility spectrum disorder in Wales, UK. BMJ Open. 2019. https://doi.org/10.1136/bmjopen-2019-031365
- Pepin MG, et al. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome. Genet Med. 2014. https://doi.org/10.1038/gim.2014.72
- Trudgian R & Flood T. An exploration of the journey to diagnosis of Ehlers-Danlos Syndrome for women living in Australia. PLoS One. 2024. https://doi.org/10.1371/journal.pone.0307574
- Medscape. Ehlers-Danlos Syndrome. https://emedicine.medscape.com/article/1114004
답글 남기기
댓글을 달기 위해서는 로그인해야합니다.